

The problem with therapeutic development is simple: we do not understand the body as a system of cells.
Researchers have spent the last 100 years studying humanity’s diverse cell types in petri dishes. Their work has largely focused on a bottom-up approach: study the individual cells and proteins to build the system.
Clinical care, on the other hand, follows a top-down approach. Clinicians are focused on the patient’s symptoms or indications and work to find the cause, and then a treatment or cure.
This disconnect is why 90% of clinical trials fail. The interactions that are obvious in a petri dish are confounded in a person, with many downstream effects that can render a promising treatment useless or antagonistic. Researchers study individual cells interacting in a controlled environment. Clinicians master complex indications burdening patients to treat their disease. The middle ground, the system, is what we do not understand.
Thus, we need a map of the human body as a system of cells to connect experimental research with clinical outcomes.
One big misunderstanding

First, an example of how incredible our cells are.
As our defensive troops, immune cells are the unit of immunity.
Immune cells patrol through our body at all times, scanning for imminent threats. Once a pathogen breaks in, they sound the alarm and nearby cells activate to meet the intruder head-on. The immune cells battle it out, eliminating the bacteria, viruses, or cancer cells, but causing collateral damage to nearby healthy cells.
Over time the immune cells neutralize the threat and reverse course, dying off to allow other cells to proliferate and repair the damaged area. The immune cell population returns to roughly the same level it was before the infection occurred within a few days. This step is essential as we only have so much energy to spare, and cannot sustain an activated immune population indefinitely. Think of how tired you are when sick.
A small fraction of immune cells, called memory cells, circulate throughout your body and remember the battle forever. These veterans memorize the identity of the intruder that caused this destruction and patrol to catch them if they ever try another ambush.
Over the course of this simplified example, immune cells determine when to attack, retreat, recover, and guard, on their own and without external input. These individual cells make split-second decisions to protect us, operating in a well-orchestrated army of troops. Dysfunction of these decisions is involved in many diseases, from ulcerative colitis to asthma to cancer.
I, along with countless other immunology researchers, analyze these decisions in a lab by culturing cells in a petri dish. We then incorporate our drug or therapy to see how it changes their interactions in a controlled environment.
This approach for understanding how immune cell populations behave is great… until we try to apply it to people.
For example, we can engineer T cells to express a receptor targeting a cancer antigen, a protein present on the surface of cancer cells but not surrounding healthy cells. In a petri dish, these CAR-T cells are excellent (CAR meaning Chimeric Antigen Receptor). The CAR-T cells annihilate tens of thousands of cancer cells in a matter of days. Replace the cancer cells with healthy ones and the CAR-T cells do nothing.
We scale these experiments up by incorporating a tumor expressing the cancer antigen in a mouse and injecting the CAR-T cells to see if the therapy improves their survival relative to a control group. For dozens of companies and research groups, their CAR-T cells have demonstrated significant improvements in the mice’s survival across many cancer types.
Despite their success in experiments and animal studies along with many clinical trials over the last 2 decades, CAR-T cell therapies have only been approved for blood cancers (specifically B-cell malignancies). The reason is twofold:
Blood cancers are easier for immune cells to target as they circulate together, continuously coming in contact with the CAR-T cells. Solid tumors are localized and fibrotic (full of fibrous proteins like collagen) that prevent infiltration.
Cancer antigens can sometimes be expressed on healthy cells in low quantities, enough to activate the CAR-T cells to target these healthy cells and leading to devastating chain reactions. Blood cancers (specifically B-cell cancers) work because we have highly specific antigens for B cells and can live for short periods without them, allowing CAR-T cells to eradicate both cancerous and healthy B cells haphazardly.
Even after teams of dozens of researchers spent thousands of hours investigating a novel therapy, the status quo procedure does a poor job at predicting the complex interactions in the human body. Petri dishes and mice are not accurate models of humans. This is true of all our cell types, not only immune cells. The decisions we make on a single-cell level across 37 trillion cells compound to form the beings we are. It has proven exceptionally difficult to recreate this complexity in an experiment or animal model.
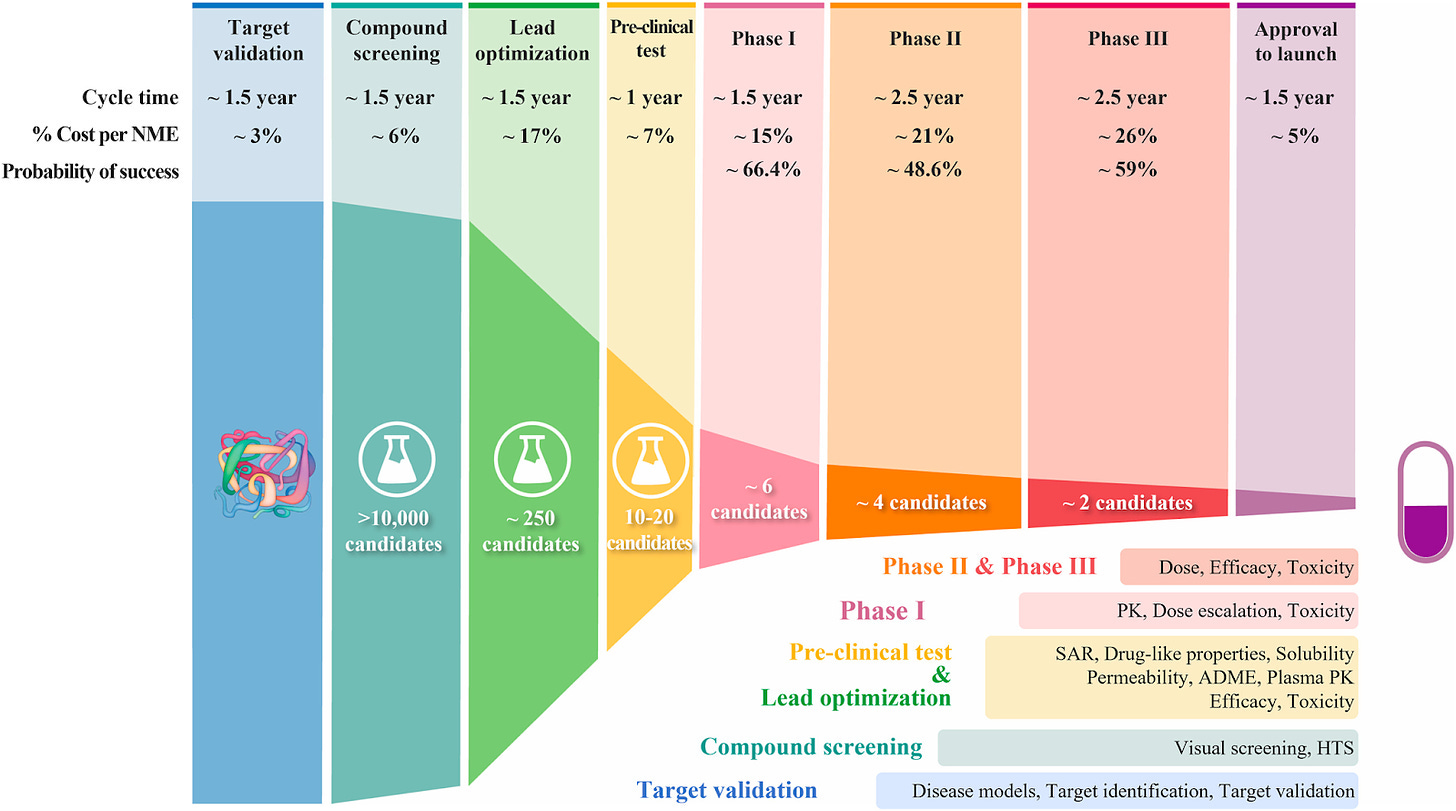
However, this approach that has powered therapeutic research and development for over a century is now complemented with a new tool to bridge this gap: artificial intelligence. Companies are combining in vitro laboratory experiments and in vivo animal studies with in silico computer simulations.
There is still something missing here, though. Many of these companies and research groups have focused their in silico simulations on the early-stages of R&D: screening thousands of drugs from a library of selections to pick a handful with the best chance to succeed experimentally to get to clinical trials. Drug screening algorithms help complement our experiments by replicating them computationally to simulate thousands or millions of potential drugs in the time it takes a scientist to test one. However, these drugs still go through the same qualification procedure in a lab and in animal models before hitting clinical trials.
We are missing a model of the human body to accurately assess if a therapy that performs well in a laboratory setting or a mouse model will translate to human patients. We need a different approach.
Biologists have uncovered how individual cells make decisions to respond to their environment. These decisions compound across tissues of millions of cells, organs of billions of cells, and eventually our bodies of trillions of cells. Maintaining the individuality of the cell’s decision-making is key here. The human body, from a mathematical perspective, is a complex adaptive system. These systems require individual agents to make decisions on local information to influence nearby neighbors in order to exhibit their extraordinary behavior at a systems level.1
We need to accurately represent these individual agents in a way that scales to the local environments of cells in tissues and organs until we can replicate the human body as a system of cells in its entirety. Doing so would provide researchers of novel treatments a way to assess their clinical potential beforehand, and would give clinicians a tool to design personalized treatment plans for patients. We need a different approach to connect research results and clinical care.
We can use artificial intelligence to model individual cells as decision-makers and map the human body as a system of computational cells.
Creating computational cells
In the early 2000s, as computers’ capabilities grew, Michael Hines and John W. Moore of Duke University poured their time into developing a computational model for how a human neuron chooses to propagate its signal to its neighbors. They devised a library of differential equations to model the activation state of each cell. Their model was tested on experimental data of neuron activation, with the goal being for the computational neurons to activate (or not activate) under the same conditions as the biological ones. This quest culminated in the NEURON model, a computer program published in 2001 and updated over time to produce one of the most accurate computational models of a human neuron.
However, this model has several caveats. Matching the accuracy of the simulated cells to biological neurons requires solving thousands of differential equations, which takes a lot of computational power and time. This is for a single neuron, too. We have ~86 billion of them in total. While differential equations can model more simplistic population dynamics, complex adaptive systems are so large one of their defining characteristics is the inability to model them with systems of differential equations.

In 2021, David Beniaguev, Idan Segev, and Michael London from the Hebrew University of Jerusalem sought to overcome this computational limitation using a new class of algorithms: deep neural networks. Recent advancements in computer optimization to train neural networks have enabled unprecedented advances in artificial intelligence. These neurobiologists trained a deep neural network to model human neuron activation and compared it with the NEURON model. Amazingly, the neural network was as accurate as the NEURON model but completed the calculations more than 2,000 times faster on the same computer [3-4].
The NEURON model and its deep neural network offspring showcase a proof-of-concept for the computational immune system map in two ways. First, we can use computer simulations to build accurate models of decision-making cells such as neurons. More importantly, we can use deep neural networks to speed up these computations without sacrificing accuracy.2
Thus, our framework for a computational human cell will be built using a deep neural network.
A simple framework
Our decision-maker is the cell. These agents are resting by default in a neutral position, maintaining our body’s homeostasis. Following provocation, cells can make decisions by changing their gene expression.
In some cases, cells will respond by expressing, or turning on, new genes that were previously dormant thereby producing new proteins. They can also silence genes that were expressed to remove the protein from their environment. There are more subtle adjustments they can make, too. Rather than turn a particular gene on or off, they can upregulate or downregulate its expression to produce more or less protein, respectively. In any case, the cells act by determining which genes to express and in what quantities.
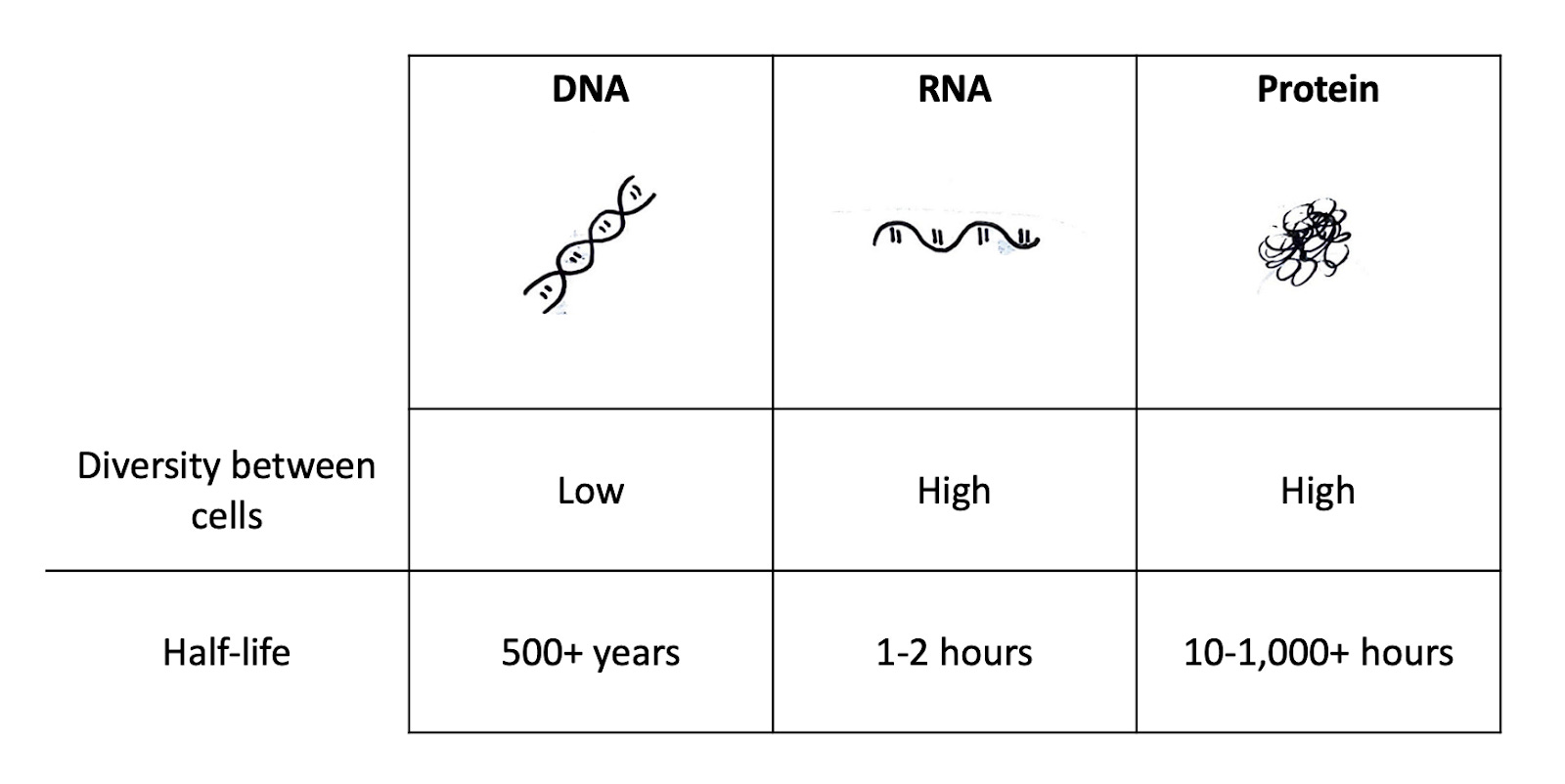
Recall the central dogma of biology: DNA→ RNA→ Proteins.
Every cell in a person has the same DNA with the same genes, but different RNA to make different proteins. Each cell chooses which DNA to transcribe into RNA and then translate into a protein. The RNA of a cell is only present for a short period, with a half-life of around 1 to 2 hours. Proteins, on the other hand, can stick around for hundreds or even thousands of hours (although this turnover rate varies dramatically by protein and cell type). Therefore, if you know the RNA inside a cell, you know what proteins the cell is currently making and thus what genes it has decided to express in response to its environment. We can measure a cell’s RNA to gain insight into its decision-making process.
Let’s dive a little deeper.
To begin, consider cell A.
Cell A is a muscle cell, also known as a myocyte. Myocytes express loads of myosin, a protein necessary for muscle contraction. Cell A thus expresses a lot of myosin, as well as other genes specific to muscle cells. This cell also expresses a lot of housekeeping genes that are necessary for all of our cells to function, such as the proteins required to fold our DNA into chromosomes.
Cell B, on the other hand, is an immune cell. It expresses the same housekeeping genes but different functional genes, such as granzymes used to kill diseased cells.
We can identify Cell A from Cell B by looking at their differences in gene expression. Further, since gene expression determines the function of a cell, we can compare the genes expressed by the same cell type across different conditions (for example, in response to different drugs) to see how their functionality changes.
So, to design our computational cellular neural network, we can input the measured gene expressions from the cell’s RNA. We can narrow down the number of possible RNA sequences to the roughly 20,000 genes in the human genome. The input of our network will have 20,000 variables; one for each gene. Since we want to know the RNA the cell will produce following environmental stimuli, the output will also be those same 20,000 variables.

We can represent this as a vector of length 20,000. Each position in that vector will represent a particular gene, with the number corresponding to its expression level.
This is called one-hot encoding. It is commonly used as a starting point for representing information numerically. While this is effective at packaging our gene expression data into an acceptable format for our neural network, the vector will be sparse, meaning many of its values are zero. Cells only express a subset of the 20,000 genes available.
We can remove unnecessary information and speed up our computations by encoding the expressed RNA sequences into a shorter vector of, say, 500 numbers called embeddings. How this happens is a little math-heavy and worthy of its own blog post3. Liam Bai recently wrote an excellent one on this exact subject.

The benefits of embedding are two-fold. First, by performing the embedding the neural network learns which genes are essential in determining cell function and which are less important. Genes that are highly variable between different cells, meaning they are expressed in very different quantities, provide a lot of insight into their differing functionality. Others that are expressed at the same level in all cells tell us far less about what the cell is doing differently.
Second, the embeddings are shorter than our original input vector of 20,000 numbers. This speeds up any computations we want to do dramatically. While our example embeddings 500 numbers long are only 40 times smaller than the input of 20,000, much of the math involved in neural networks is matrix multiplication. This means the computations are 500x500 matrices, with 250,000 individual numbers. Substantially smaller than the 400,000,000 numbers in a 20,000x20,000 matrix. At the end of our calculation, we are able to recover our 20,000-long gene expression vector from our embeddings using a decoder. Our embeddings allow us to do more with less.
Let’s discuss how we incorporate our environment into the calculation (hint: it’s by creating another embedding). We have our base state of gene expression as our input, representing cells behaving as they normally do. We then introduce factors such as therapeutic drugs and measure the cell’s gene expression to compare the cell’s original state with this new augmentation. We train the network to learn how to represent these environmental factors to match the change in gene expression between the control cells and the cells that were given the drug.
In other words, we have our embeddings of a normal cell with no treatment and another of a cell treated with some drug. We ask the neural network to find an embedding of the environment that, when multiplied with the normal cell’s embeddings, produces the treated cell’s embeddings.
The broad structure of the model is as follows.

So we have our representation of gene expression, and we have defined our input as the current genes expressed in the cell with the output being the genes the cell will produce next. In the middle, the environment influences how the cell chooses which genes to express.
This network design is a novel deep-learning structure out of Fabian Theis’ lab at the Helmholtz Munich Institute of Computational Biology. Published in 2023 and called the compositional perturbation autoencoder, this framework can predict the response of many cell types to novel combinations of drugs [5]. It models a single cell as a set of gene expressions and predicts their change in response to completely unseen environments. These are our computational cells.
We still need to scale these decision-makers to the whole system. To get there, let’s first discuss the data needed to teach these individual cells.
Putting it to work
To teach our computational cells how to behave, we need to know how actual cells behave: our ground truth. A growing suite of experiments designed to assess how single cells function is providing this data to feed into our computational cellular model.
Biotechnology is going through a sequencing golden age. Sequencing refers to experiments designed to measure the order of DNA or RNA nucleotides, the alphabet of our genes. The cost of sequencing DNA and RNA has shrunk 10,000-fold over the last 2 decades.

More recently, researchers have developed ways to sequence the RNA of a single cell4, enabling us to understand how these individual agents behave in a laboratory or clinical setting. This technology has already unraveled the diversity among cell types that were previously thought to be identical, including immune cells in our brains. Single-cell RNA sequencing data is exactly what we need to measure the gene expression levels of our computational cells.
We can take single-cell RNA sequencing data from patients before and after treatment to understand how the drug impacted cellular decision-making and compare it to results from lab experiments to see how they differ. With this data, we can isolate different cellular populations to see how a drug designed to target muscle cells impacts off-target cells in the lungs, for example.
Over time, this model will build a library of decision-making cells to represent the hundreds of unique cell types in our body.
As our model becomes more accurate at predicting cellular behavior in patients, we can use it to infer how experimental results will translate to a clinical setting for novel treatments and bridge the gap between research and clinical care. Even modeling a single cell will provide invaluable insight into healthcare.

Single-cell sequencing technologies are paramount to this vision. The agents of the human body are individual cells, with each displaying unique functionality and decision-making. Building a machine learning-powered model of these systems requires data on these cell’s decision-making. For the first time ever, we are able to generate this data (and some research groups have begun posting it!). Now we can put it to use for something truly monumental.
Okay, so we have our computational cells. How do we build a system of cells?
{kind=link}
In a hospital one of the most essential tools for diagnosing patients is histopathology. A pathologist will take a tissue sample from the organ of interest, create a layer one-cell thick, place it on a slide, and analyze the cells for abnormal patterns. To identify distinct cell populations, the pathologist will stain the slide for particular proteins. The slides uncover the diverse populations and structure of a tissue at a cellular level.
We can use these histopathology slides to model microenvironments where thousands of diseased and healthy cells of many different cell types interact with one another. Using computer vision algorithms, we can translate the patient slides into a computerized map of cells and use RNA sequencing data to figure out which cell is what type (e.g., a muscle cell from an immune cell). From there we insert our computational cells in place of the various cell types to analyze how they behave. We now have a small environment of cells interacting with one another representative of a patient’s tissue. This allows us to validate the accuracy of our single-cell model in microenvironments of thousands of cells.
Moving forward, we can input drugs or therapies to see how this impacts the cells’ decision-making and the expression of genes throughout the microenvironment. Histopathology slides and RNA sequencing taken before, during, and after treatment will monitor the progress a patient makes and the drug’s impact on gene expression to assess how well our model predicts cellular behavior on a larger scale.
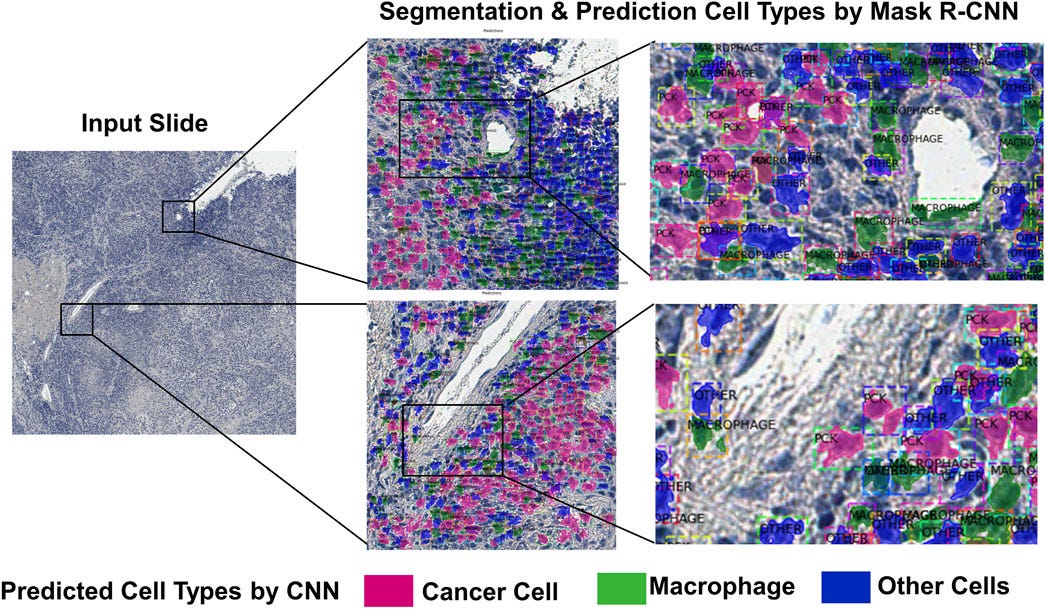
With a validated model of computational tissues of single cells, clinicians and researchers can predict the effect of novel treatments on the disease microenvironment before prescribing any medication to maximize the patient’s chance of recovery. They can combine therapies or adjust their dosage to fit the patient’s exact needs by trying many different possibilities in the model beforehand. Our computational cellular model applied to patient’s clinical data is key to creating personalized therapies.
To grow our map further, we can take slides and RNA from several distinct tissues, such as the tumor, the associated organ it’s occupying, and nearby lymph nodes for a cancer patient. We can track the cells’ expression levels throughout these areas to see how a drug targeting the tumor impacts the surrounding healthy organ cells and the immune cell populations in the lymph nodes. The model could predict how a subset of cancer cells may evade the drug and provide a second treatment option in advance to eradicate those cells.
Over the course of treatment, the program monitors the expression of genes by individual cells and uses clinical histopathology slides to build out our map, connecting the communication lines of cells between the tumor, organ, and lymph nodes. From there, pathologists and clinicians can see which treatments to prescribe that will be most effective at overcoming the disease.
Our single computational cell allows us to complete a map of how these cells come together to form tissues across organ systems, and how treatments impact this organization of cellular decision-making. We build our map by analyzing these changes in gene expression on the single-cell level and grow in scale to our entire body.
Caveats and considerations
One key limitation of sequencing the DNA or RNA of a cell is it is destroyed to extract its nucleic acids. This means we cannot sequence the same exact cell across multiple time points; before and after a treatment, for example. Mathematicians and scientists have designed workarounds called optimal transport algorithms to predict the genes a cell is likely to express at a future time point. There are some issues with this approach, though, including its high computational complexity, making it infeasible for large models like the one proposed here.
However, another option may be to just throw enough data and computational power at the model and let the neural network figure it out. Artificial intelligence in the form of language models went through many clever redesigns before the landmark paper in 2017 created the transformer model. These transformers were so good at picking apart the necessary components from a sequence of text, they kicked off an arms race of “who can train the biggest language model”. This culminated in the chatbot known as ChatGPT which popularized generative artificial intelligence.
You may be asking yourself “Yeah, that worked for ChatGPT, but will it work for a biological cell?”. A fair question. In 2019 a research team at Meta published a large language model called ESM that predicts the 3D structure of a protein by its sequence. They found that adding more data and computing power to the model increased its ability to recognize biological patterns in protein sequences. Perhaps the most eloquent way to improve our single-cell sequencing model is to learn from ChatGPT and ESM: use a transformer to generate our gene expression embeddings and torrent it with sequencing data and computer hardware until it picks apart the optimal paths for each cell to take. Adam Green, the founder of Markov Bio, penned an essay on this exact rationale.

Consider a single cell. The human body contains around 30 trillion cells. Modeling one cell really well is great, but it is far from capable of producing system-level complexity if we cannot scale it.
If we assume it will take at least 50 million parameters to model a single cell:

Then our human scale model will require a minimum of 1.5 sextillion parameters.
The scale of this proposed model is large, don’t get me wrong, but perhaps not infeasible. Consider OpenAI’s ChatGPT model, which they publicly state has 175 billion parameters and some software engineers postulate it has been upgraded to over a trillion parameters. Only 5 years earlier their top-end model had 117 million parameters. Computer hardware and, perhaps more importantly, software to train machine learning models continues to accelerate.
So, a model of the entire human body is unfathomably large by today’s standards but not out of the question for computers 5 or 10 years from now. A model of a single cell is within our grasp, and even that is enough to uncover avenues for improving research and patient care. Besides, along the way, we will be inundated with models of local microenvironments of tissues that will prove invaluable to our understanding of countless diseases. The journey is as important as our destination.
Predicting the future
We can consider this project progressing in 3 stages:
Model a single cell really well.
Build our library of models for the many cell types of the human body.
Grow in scale beyond a single cell to model local tissues, whole organs, and organ systems, until we map the entire computational human body.
We are currently at step 0 in modeling the human body. Before we can, we first have to model a single cell. Researchers are racing to develop the assays and data needed to model cells and their decision-making. What we need is something groundbreaking to put all that data towards, and that is exactly what I am proposing.
Progress will inevitably come in small wins: modeling the activation choices of immune cells; differentiating between the muscle cells of young and elderly people; describing the gene expression from healthy to precancerous to cancerous cells. The exact path we take is shrouded ahead of us. The goal remains the same: model computational single cells and scale to a complete map of the human body.
The year is 2119 and a novel coronavirus emerges with rapid infection potential. Having learned from the last pandemic a century prior and with new tools at their disposal, clinicians immediately take histopathology slides, blood samples, RNA sequencing, and CT scans to analyze the immune response of these initial patients. The healthcare workers look at the data themselves but also feed it into a computational human map. The mechanism for its infection and distribution throughout the body is broadcast immediately on these patient’s maps. The clinicians present can evaluate different drugs already available in the hospital to treat these existing patients, while researchers analyze their gene expression to identify disease-specific targets for new drugs. Patients are given personalized treatment regimens for their unique conditions. The map removes the guessing game of determining treatment, existing or new, for a novel disease mechanism. This is all completed in real-time before millions of people are infected and the globe is shut down.
A platform for personalized insight into patients’ cells provides invaluable perspective to researchers designing precision treatments and physicians making life-saving decisions. It is the glue connecting bottom-up and top-down approaches to healthcare.
This is a moonshot idea. Mapping the 30-trillion-cell human body as a system is a task on par with sequencing the human genome in the 1990s; at the time, scientists questioned if it would even be possible. Its implications are enormous, too. Imagine the success rate of clinical trials increasing from 10% to 20%-25% or beyond. The pace of innovation would accelerate and, simultaneously, the cost of new medicines would plummet. It’s clear we can accomplish this monumental project by starting with the resources we have today and growing in scale as they advance. A human body map would form the bridge between laboratory research and clinical care for the betterment of all humanity.
We can map our way to better healthcare for all.
Thank you for reading Positive Selection! To receive these newsletters directly in your email inbox, subscribe for more.
Appendix:
For a visual example of complex adaptive systems, I encourage you to watch this murmuration of starlings. Each starling follows the behavior of their closest 7 neighbors, and without any clear leader, the flock exhibits extraordinary flight patterns.
When scaling systems’ simulations to large populations, any decision by a single cell compound in the larger system, and the mistakes of those decisions will compound, too. This is known as the propagation of uncertainty. Thus it is essential to model individual agents as accurately as possible to minimize these errors.
In essence, we start with a neural network input layer of our 20,000-length vector of genes. We then transform this to our embedding layer via an encoder. The encoder’s task is to find a way to represent these 20,000 numbers in a shorter length we specify, such as 500, without losing out on any key information. We can use this embedding layer to simulate our environment, incorporating factors like nearby proteins produced by other cells or therapeutic drugs to change the embedding layer. As I mentioned earlier, the benefit of the embedding is that computations can be completed much faster, which is important for scaling to a system of many cells. The drawback is we lose out on context. Each position no longer corresponds to a distinct gene; this is what is referred to as the “black box” of machine learning. Thus, we use a decoder to recover our 20,000-long gene expression vector from the embedding to extract the information we desire: what genes the cell expresses afterward.
We can teach our neural network how to accurately represent our gene expressions as embeddings by leaving out the environmental stimulus. The cells should thus express the same exact genes at the input and output of the network. This model is called an autoencoder. It takes our initial gene expression vector, encodes it into a shorter vector for easier mathematical operations, and recovers the initial sequence by decoding it to ensure the representation is correct. To learn, we feed the autoencoder sequencing data from cells and attempt to encode and decode the same sequence. The first time around, the embeddings are terrible and the original vector cannot be recovered. However, the algorithm learns from its mistakes and improves over time, eventually being able to encode the 20,000-long gene expression vector into an embedding, and then decode it to recover the same vector. By doing this the autoencoder learns how to represent the gene expression vector as a shorter embedding without losing any key information.
Single-cell sequencing works by isolating a single cell in a dish, extracting its DNA or RNA, and sequencing it. There are some considerations to this workflow. A single cell has an infinitesimal amount of DNA or RNA; while a standard sequencing run will extract the nucleic acids from millions of cells, single-cell sequencing is limited to picograms of starting material. This starting material needs to be copied using polymerase chain reactions (PCR) to exponentially amplify each piece. All of this compounds into many error-prone steps that can augment the final data born from this assay. However, for all of its faults, single-cell sequencing has already identified dozens of novel cell sub-populations that were hiding in plain sight. One example of such a discovery is a subpopulation of immune cells in the human brain. The protocols and assays used to generate this groundbreaking data will continue to improve over time.
References:
[1] Wong CH, Siah KW, Lo AW. Estimation of clinical trial success rates and related parameters. Biostatistics. 2019;20(2):273-286. doi:10.1093/biostatistics/kxx069
[2] Sun D, Gao W, Hu H, Zhou S. Why 90% of clinical drug development fails and how to improve it? Acta Pharmaceutica Sinica B. 2022;12(7):3049-3062. doi:10.1016/j.apsb.2022.02.002
[3] Beniaguev D, Segev I, London M. Single cortical neurons as deep artificial neural networks. Neuron. 2021;109(17):2727-2739.e3. doi:10.1016/j.neuron.2021.07.002
[4] Hay E, Hill S, Schürmann F, Markram H, Segev I. Models of Neocortical Layer 5b Pyramidal Cells Capturing a Wide Range of Dendritic and Perisomatic Active Properties. PLOS Computational Biology. 2011;7(7):e1002107. doi:10.1371/journal.pcbi.1002107
[5] Mohammad L, Anna KS, Carlo DS, Leon H, Yuge J, Ignacio LI, Sanjay RS, Mohsen N, Riza MD, Beth M, Jay S, Jose LMF, Pierre B, F Alexander W, Nafissa Y, Stephan G, Cole T, David LP, Fabian JT. Predicting cellular responses to complex perturbations in high-throughput screens. Mol Syst Biol. 2023;19:e11517. https://doi.org/10.15252/msb.202211517
[6] Triana S, Vonficht D, Jopp-Saile L, et al. Single-cell proteo-genomic reference maps of the hematopoietic system enable the purification and massive profiling of precisely defined cell states. Nat Immunol. 2021;22(12):1577-1589. doi:10.1038/s41590-021-01059-0
[7] Lee K, Lockhart JH, Xie M, Chaudhary R, Slebos RJC, Flores ER, Chung CH, Tan AC. Deep Learning of Histopathology Images at the Single Cell Level. Frontiers in Artificial Intelligence. 2021;4. https://doi.org/10.3389/frai.2021.754641
[8] Epoch, "Parameter, Compute and Data Trends in Machine Learning". Published online at epochai.org. Retrieved from: https://epochai.org/mlinputs/visualization [online resource]
People have been throwing around ideas like this for years in some incanrnation or another. This is just a rehash of these old ideas updated in an “AI” cloak. It’s noble for sure but it’s not practical because of everything we don’t know.