
Transcription: Decoding Cancer
The Human Tumor Atlas Network is doing this by analyzing the thousands of proteins that individual cancer cells produce.

Hello, and welcome to Transcription, a weekly overview of research happenings in biotechnology and immunology! Subscribe to stay informed and support my work:
Cancer is an artifact of evolution’s incessant march forward. Performance-enhancing mutations in cells’ DNA, either passed along via heritability or as a result of environmental factors, enables cancer to bypass the biological rules of the game. These cells can overcome mitotic checkpoints to continue dividing indefinitely, downregulate communication molecules to avoid immune responses, or recruit restorative stem cells to assist in their growth. And the societal impact of cancer cannot be overstated. Cancer is the second leading cause of death in the world. We need to figure out how to treat this devastating class of disease.
The conventional wisdom behind treating cancer revolves around identifying what makes cancer cells different from healthy ones. And not just from the perspective of a trained doctor. In most cases once cancer becomes noticeable it has spread far beyond what a surgeon can remove from the patient. What we need is a biomolecular way to distinguish cancer from health. In other words, a drug. We need to design a drug that can tell cancerous from noncancerous.
Ultimately, cancer is a personal disease. And this makes the task of treating cancer more difficult. We are all individuals, down to our DNA, and cancers are no different. There are groups of cancers with similar treatment plans, ascertained from centuries of trial-and-error. But ultimately an oncology team for a patient is tasked with a unique challenge. And understanding the difference between an individual’s cancer from their healthy cells is essential to developing a successful treatment plan.
The Cancer Moonshot initiative began in 2016. In 2018 it started a program to develop an atlas of human tumors (the aptly named Human Tumor Atlas Network). In essence, build a collection of maps for how cancer exists in three-dimensions to help us understand how to better diagnose and treat patients. At the end of October they published a series of papers detailing their latest results. What they published and continue to research is destined to change our understanding of cancer: how cancer originates, how malignant disease progresses, how cancers resist treatment, and how to effectively cure patients.
Going through the 12 papers they published at once is a bit much for a weekly newsletter. I would rather sink my teeth into them and focus on key takeaways week-by-week. These are some of the most interesting and impactful findings in the field of cancer biology and immunology of the year, and I want to do them justice.
And I think the natural starting point is understanding what makes such groundbreaking research possible at this moment in scientific history.

Over the last century there has been a paradigm shift in understanding what cancer is. And a huge part of this has been improving the tools available to clinicians and scientists studying cancer. Historical diagnostics and treatments relied on identifying malignant physical lumps on patients. Advancements in microscopes and surgical techniques enabled researchers to study cellular abnormalities in the 19th century. Post-war scientists studied how derivatives of mustard gas stop cell growth and developed the first modern chemotherapy drugs. Aseptic cell culture of cancer cells like Henrietta Lacks' cervical cancer allowed scientists to try to treat cancer cells outside of patients. Chemotherapies could be tested in labs and animal experiments before moving to clinical trials, creating a diverse crop of drugs available for the dozens of cancer types identified to date.
Today, though, we have automated machines and high-throughput methods that can isolate the components of individual cells, scanning through millions of cancerous and healthy (stromal) cells in patients. These new methods let us uncover those biomolecular differences between healthy and cancerous cells with higher resolution.
What this new crop of data has uncovered is the diversity of cancer. Not just patient-to-patient, but within a single patient’s tumor. Cancer is not a homogenous crop of a single cell type gone awry. It's a concert of many cancer cell types working with stromal cells to form a new organ. This cancer organ requests and receives new blood vessels from the cardiovascular system, and communicates with nearby cells to help clear debris.
Treating cancer as a homogenous ball of cells was necessary because we couldn’t actually tell the difference between the components of the tumor. Our diagnostic tools relied on looking at the shape of cancer cells, or at looking for the expression of a single protein. And ultimately that led to therapies targeting individual proteins commonly seen in cancers, like PD-L1, VEGF, and HER2. Each of these targets now has antibodies targeting them that rake in billions of dollars each year.
But we have essentially run out of targets, more or less. There are very few tumor-associated antigens that can effectively cover a majority of cancer cells without also damaging healthy counterparts. And those have all been drugged in some sense, with monoclonal antibodies or small molecules. Yet I believe this is a huge oversight given the data we now have our hands on.
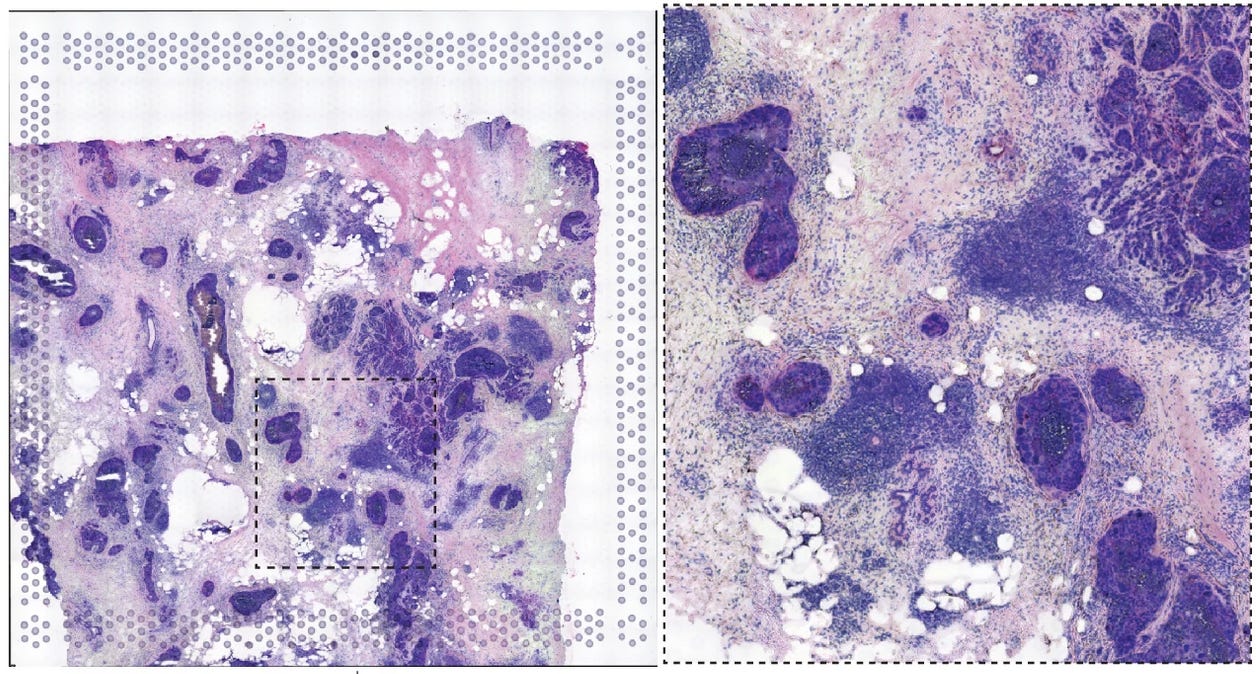
Cancer is diverse! Chemotherapy regimens are rarely just one drug as a result, combining several treatments into a cocktail. And indeed protein-targeting therapies like HER2-antibodies are often used as a component of a patient’s therapy that includes other drugs. Even for patients with cancer that expresses a lot of HER2 protein, that one therapy is not enough to get all of the cancer cells. And while HER2 antibodies are approved for breast cancer patients, they have not been successful at treating HER2+ ovarian cancer. Both cancer types express the HER2 protein, but one of them is treatable while the other is not. What gives?
Think of antibiotic resistant bacteria. The liberal use of antibiotics like penicillin saved many patients’ lives who had bacterial infections like pneumonia or tuberculosis. But extending that use further to agriculture and to non-bacterial infections enables bacteria to evolve for antibiotic-resistance. Eventually, by chance, a bacteria will evolve that is unaffected by the antibiotic. And that antibiotic becomes useless as the bacteria divides indefinitely. And new antibiotics need to be discovered to treat the previously antibiotic-resistant bacterial infections. This game of cat-and-mouse goes on-and-on.
The same is true of cancer. Using a single protein target may be able to get most of the cancer cells. But some of them by chance may not express that protein. And after treatment, those targetless cells will avoid treatment, recover and the cancer will relapse. This time the patient needs a new treatment, as the current malignant disease is resistant to the previous therapy.
So even for targeted therapies like monoclonal antibodies targeting cancer proteins, they are combined with chemotherapies for complete eradication of the cancer cells. And those combinations are certainly better than the antibody alone or the chemotherapy alone. But they are far from perfect, with many side-effects from off-target toxicities. And even with these advanced therapies, the death rates for most cancer types have not improved in 30 years.
What we need are more targets. Rather than looking at single proteins to see which are highly expressed in cancer cells, we need to see which combinations of proteins are identifiable on cancer cells. This makes therapies more resistant to being out-competed by cancer’s hyper evolution.
The Human Tumor Atlas Network is doing this by analyzing the thousands of proteins that individual cancer cells produce. That way they can look at treatment targets combinatorially. Maybe there isn’t a single protein that targets all cancer cells, but co-expression of two or three proteins can cover them all.
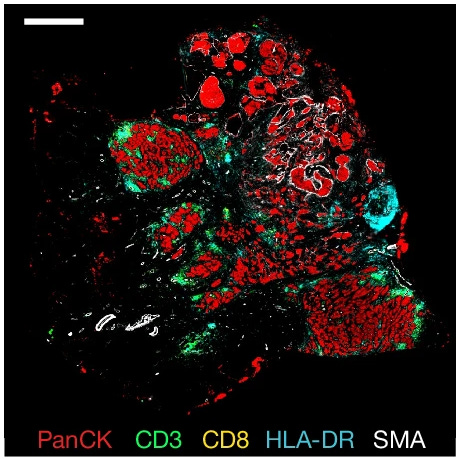
The cell-staining methods of old have been replaced by techniques like CODEX that look at 60-proteins expressed in a single cell. Or spatial transcriptomics that can analyze which genes are expressed in individual cells while preserving their place in the tumor tissue. Or combine them both, and get a map of a patient's tumor with the proteins and RNA expressed in individual cells. In total, 6 of the papers published by the HTAN consortium detail using these multi-modal methods to analyze individual cancer cells in patient’s tumors. Another 5 papers develop new methods for generating this data or analyzing it computationally.
What these new assays provide is an immense amount of data. Data that can (and is) fed into large AI models to help pick-apart the differences between patient samples and between cancerous and healthy cells. These algorithms can help researchers and clinicians figure out which combinations of protein targets effectively cover the widest swath of cancer cells while leaving the healthy cells untouched.
These methods are novel, with some only being developed in 2021. This, in tandem with the advancement of artificial intelligence systems, has enabled single-cell profiling of cancer cells to design next-generation therapies.
The diversity of cancer has been noted for decades, with theories for how diverse cancer cells occur within a single patient dating back to at least the 1970s. But studying this diversity with previous state-of-the-art laboratory equipment has been slow and cumbersome. In the last decade, experimental methods have caught up to enable us to study individual cancer cells. And computational tools have similarly advanced, allowing us to pick apart from this immense data source how to create new treatments. This is the right place and time in history for a cancer moonshot, and we are poised to take advantage of this in the coming decades.