

By far and away the biggest scientific news of the past week was the announcement of 2024’s Nobel Prize winners. So let’s dive into why these scientists won.
What the hell is “microRNA”?
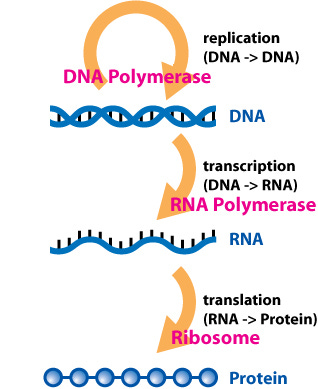
Our cells all have the same DNA. The same instruction set that tells them which proteins to produce. The 20,000 or so genes throughout our DNA each encode for different RNA molecules that instruct our ribosomes to conjoin a string of amino acids that will produce the final protein. The proteins are what ultimately allow our cells to function. And, ultimately, the different proteins in our cells differentiates, for example, a muscle cell from a blood cell. A muscle cell will be full of myosin, which allows them to contract in response to calcium signals from nearby neurons, while blood cells are instead pumped up with hemoglobin to carry oxygen throughout our blood stream.
{kind=link}
The challenge is, how does a muscle cell know to produce myosin and not hemoglobin, and vice versa for the blood cell? They are given the same exact instruction set in their DNA, after all.
This is the foundation of epigenetics. Our cells have the same sequence of DNA, but there are modifications on top of that DNA that controls which genes are produced in different cell types. These are epigenetic modifications that do not change the sequence of DNA. Genetic modifications, in contrast, change the sequence of DNA (the A, C, G, and T that encode our RNA and protein sequences).
An example of epigenetic modification is adding a methyl group to DNA, changing the chemical properties of that region of DNA and enhancing (or suppressing, in some cases) its expression into RNA. Another is the binding of transcription factors to DNA. Transcription factors are a class of protein that bind to DNA regions and turn on or off gene expression. These modifications do not change the sequence of DNA. In other words, they do not alter the information it encodes. But they change the amount of RNA that is produced from these regions of DNA.
In both cases, these modifications change the biochemistry of RNA polymerase, the enzyme that transcribes DNA into RNA. Sometimes they help RNA polymerase move along the DNA strand and make more RNA, other times they stop RNA polymerase and suppress DNA to RNA transcription in that particular gene.
But this is all occurring at the DNA level. These epigenetic modifications are changing the biochemistry of DNA molecules only, affecting the transcription of DNA to RNA. This process is commonly referred to as transcriptional regulation.
And it turns out that transcriptional regulation is not enough to explain the difference of gene expression between cell types.
In 1992, Victor Ambros’ lab at Harvard University discovered that nematodes known as C. elegans have two reciprocally expressed genes. The gene lin-14 is essential to early development of the small worms in their larval stage. Once these animals progress beyond larvae, lin-14 protein levels rapidly decline. What Ambros’ lab found was that the lin-14 RNA molecules are still present, meaning the gene is still being transcribed from DNA into RNA. But they form a duplex with a second RNA molecule from the gene lin-4. The RNA of lin-14, which are messengerRNA (mRNA) molecules that encode a functional protein, are attacked by lin-4 microRNA. As the name suggests, the lin-4 RNA molecule is very small. While the lin-14 mRNA molecule is around 1,600 base pairs long, the lin-4 microRNA 22 base pairs long. Yet it is still enough to stop the lin-14 mRNA from working properly.
Ambros’ colleague Gary Ruvkun, also at Harvard Medical School, showed that the binding of lin-4 microRNA to lin-14 mRNA prevents ribosomes from passing along the mRNA molecule to form the final protein. The microRNA forms three-dimensional bulges that prevent the ribosome from progressing through the molecule. The ribosomes are ultimately ejected from the mRNA, which means that the lin-14 protein is not fully produced and thus not functional.
This class of gene regulation is called post-transcriptional regulation. The RNA polymerase was able to transcribe the DNA gene into mRNA. But this regulatory mechanism prevents the mRNA molecule from ultimately translating into a functional protein.
Ambros’ and Ruvkun’s work showed a novel class of RNA molecules, microRNAs, that are essential to C. elegans development. Yet the following year, Victor Ambros was denied tenure at Harvard, as they did not believe this discovery was significant enough for him to remain a member of their faculty. Scientists were skeptical that microRNAs were relevant to biology in general and believed that they were limited to the development of nematodes. There were no other papers published on microRNAs until the turn of the millenia, seven years after their initial discovery.
What changed was the discovery of the second microRNA. Also in C. elegans, the Ruvkun lab discovered lin-7, a microRNA that suppresses the translation of lin-41 in a very similar fashion to lin-4’s regulation of lin-14. The significance of this discovery was that lin-7 is also present in other genomes across the animal kingdom, including humans. Suddenly human scientists caught on that microRNAs could impact human development, and they started to care about microRNAs.
From 1993 to 1999, there were 0 additional papers on microRNA. In 2000, there were 5 papers published on microRNAs. By 2010, there were nearly 4,000. And in 2020, 19,803 papers were published with data on microRNAs.
This past week in 2024, Gary Ruvkun and Victor Ambos shared the Nobel Prize in Physiology or Medicine. A full 32 years after their initial discovery, and 31 years after Victor Ambos was denied tenure at Harvard for this very discovery being “insignificant”. Now microRNAs are being studied for their role in diseases including diabetes, cancer, Alzheimer’s, alcoholism, and heart disease.
There is something remarkable about discoveries going so against the grain of conventional wisdom that it takes the scientific community decades to recognize its significance. Because in many ways that is the point of science! Hypotheses are meant to challenge the status quo, to go against what the community believes to be true, in search of what is objectively true. I am reminded of Nicolas Copernicus and Galileo Galilei, who were met with intense criticism and house arrest for suggesting the Earth was not the center of the universe. It took hundreds of years for that theory to become widely accepted. Hopefully we as a society can correct our biases quicker in search of the truth. In this case Victor Ambos gets the last laugh, championing his Nobel Prize winning work in his lab at Harvard’s local competitor, the University of Massachusetts.
Predicting protein shapes from X-rays machine learning
Proteins form the structure of our cells. They surveil from the surface of cells, communicating with their neighbors and passing along environmental signals to the interior of the cell. Proteins catalyze reactions as enzymes. They regulate gene expression as transcription factors. So on and so forth. Everything a cell needs to function is carried out by proteins.
The function of proteins is determined by their structure. Function follows form, in this case.
We just talked about how the different expression of DNA genes into RNA and subsequently proteins makes the difference between a muscle cell and a blood cell, or a neuron and an immune cell. The DNA encodes the information needed to build the protein, and the RNA tells the cell’s ribosomes which proteins to make (and how many of them). But it's the proteins themselves that ultimately do what the cell needs them to.
And unlike DNA or RNA, proteins form really bizarre shapes based on their sequence of amino acids. Broadly speaking, DNA has the same physical structure no matter what its sequence is. The DNA will form its famous double helix. RNA is mostly the same, although its structure is somewhat less consistent. Some RNA molecules can bulge and fold back on itself, forming hairpin structures that can impact their functionality. The microRNA molecules we just talked about use these bulging three-dimensional structures to stop ribosomes from translating mRNA molecules into proteins, for example.
But proteins operate on another level of complexity.
The information contained in the gene of an orderly piece of DNA is transcribed into a mostly linear strand of mRNA inside the nucleus of a cell. That mRNA, barring any interfering microRNAs, leaves the nucleus and binds to several ribosomes. The ribosomes themselves are large structures composed of dozens of proteins that come together and sandwich the mRNA molecule. Traversing along, the ribosome builds a string of amino acids encoded in the mRNA molecule.
As soon as that string of amino acids is formed, it immediately folds in on itself. While DNA and RNA are both negatively charged no matter what their sequence is, different amino acids in the sequence of a protein have different biochemical properties. Some, like glutamate or aspartate, are negatively charged, while others like arginine or lysine are positively charged. Negative and positive charges attract one another, causing these amino acids to want to be close to each other. But also some amino acids are hydrophobic, meaning they are afraid of water. So they want to be shielded from the water inside of a cell by hydrophilic amino acids who love it.
Protein folding is so complex in fact that there are whole systems of organelles within our cells dedicated to helping proteins fold properly and transporting them to their destination. There are so many factors that influence how a protein folds in on itself. And its final form, the folded protein, is ultimately what determines its function! So we really want to know what different protein sequences look like to understand how they function. But we cannot figure out how a protein’s amino acid sequence determines its form. It’s too complex.
For nearly a century, the most efficient method for determining a protein’s structure was X-ray crystallography. In the early 1910’s, Max von Laue showed that X-rays, then only recently discovered, cause diffractions when shined through a material based on that material's chemical structure. In essence you shine an X-ray through the material you want to test, and catch the X-ray diffractions on a film photography-like material called a photographic plate.

{kind=link}
Right. So what does this abstract artwork tell us?
Like shining white light through a prism, the resulting rainbow of colors tells us what is hiding in the white light.
Measuring the angle between the diffractions, as well as their relative intensity on the photographic plate, enables crystallographers to reverse-engineer the structure of electrons needed to create that pattern. These researchers iterate on these structures, using different X-ray wavelengths and different purification methods to prepare their material as cleanly as possible.
This process is intense. It takes many iterations to solve the crystal structure of a molecule. And biochemical molecules, particularly proteins, are super complex. Copper sulfate (CuSO4) contains 6 atoms per molecule. A protein commonly contains tens of thousands.

{kind=link}
Despite the rigorous trial-and-error of determining a protein’s structure by X-ray crystallography, it still remains the most effective and efficient method for figuring out how proteins fold. Many PhD theses of the last century were dedicated to finding the structure of various biochemical molecules and proteins.
And for good reason. At least 14 Nobel Prizes were given to a total of 29 recipients relating to X-ray crystallography, either for developing it or for utilizing it to determine the structures of various proteins. Notably, Rosalind Franklin used X-ray crystallography to prove the double helix shape of DNA for Francis Crick and James Watson. And Dorothy Hodgkin discovered the structure of numerous essential biochemical molecules, including penicillin, insulin, and vitamin B-12, which won her a Nobel Prize in 1964. Indeed the eldest Nobel Prize went to Max von Laue for discovering X-ray crystallography in 1914, while the newest was awarded to Brian Kobilka for studying G-protein-coupled receptors with X-ray crystallography and other methods in 2012. This is truly a timeless experimental method.
Yet the 2024 Nobel Prize of Chemistry went to researchers who seek to replace X-ray crystallography with machine learning.
For good reason. Despite its profound success, X-ray crystallography is a process that can take years or even decades to solve the structure of a single protein. Dorothy Hodgkin spent thirty years trying to solve the structure of insulin, which she successfully did in 1969, five years after winning the Nobel Prize (beast).
So in 2010 Demis Hassabis founded DeepMind, with the initial focus of developing artificial intelligence in the context of video games. That shifted in 2016, when the team began to focus on solving the tedious protein folding protein. Hassabis hired John Jumper, whose PhD thesis focused on new methods of predicting protein structures. In 2018 they showcased their early model, AlphaFold, which won the Critical Assessment of Structured Prediction (CASP) for being the most accurate protein folding prediction tool.
In 2020, they brought their second variant of the program, known as AlphaFold2. AlphaFold2 won the CASP yet again. But with their improved model, the protein structure predictions were so accurate they could replace X-ray diffraction. The predictions from that model were as accurate as most experimental X-ray crystallography results. Rather than perform painstaking crystallography experiments over years, researchers could now test the structure of proteins in a matter of seconds. Just by giving an algorithm the protein’s sequence of amino acids.
The Nobel Prize was shared with David Baker, a professor whose lab developed its own artificial intelligence algorithm (RoseTTAFold) to predict protein folding. Baker used these tools to design completely new proteins from the ground up with novel properties. He envisioned that we could use these predictive tools to tell an algorithm “we want a protein that folds this particular way,” and the algorithm will produce the amino acid sequence to achieve that folded structure.
This vision culminated in the design of a novel protein to neutralize the SARS-Cov-2 virus that causes Covid-19. These tools enabled researchers to rapidly understand the structure of the virus, note the spike protein it uses to enter our cells, and design a protein to counteract it. And to do so quickly, not over the course of decades but in a matter of weeks.
We are entering a new era. Many pharmaceutical compounds that we use to treat diseases come from nature, and are used as-is or slightly modified before treating human diseases. Nature is the best inspiration. Now, these machine learning tools can study hundreds of thousands of natural protein sequences and their resulting structures to design completely novel ones.
This would all not be possible without the countless hours dedicated to X-ray crystallography over the last 100 years. The AlphaFold2 algorithm was trained on more than 170,000 experimentally determined structures, which researchers selflessly made available to the public domain for free. Let’s hope, for humanity’s sake, that they return the favor and keep the AlphaFold algorithms free and open to the public.
Are artificial neural networks really a physics discovery?
By far and away the most controversial scientific Nobel Prize of 2024 was the physics award. Not because the discovery was insignificant - in fact it enabled the Nobel Prize in Chemistry! - but because it’s debatably not in the field of physics at all.

Artificial intelligence has exploded in public interest over the last few years, particularly since the launch of ChatGPT. But the theory behind AI has been under development for decades. In the 1940’s, Warren McCulloch and Walter Pitts invented the perceptron, a simplistic machine learning algorithm based on the individual neurons in our brain. This theoretical “artificial neural network” was implemented in the Mark I Perceptron machine in 1957. The US Navy hyped up the media about the machine’s capabilities, particularly relating to machine translation (algorithmically translating one language into another; think Google Translate). And it was then demonstrated in 1960:
"the embryo of an electronic computer that [the Navy] expects will be able to walk, talk, see, write, reproduce itself and be conscious of its existence." - The New York Times, 1958
The demonstration was a complete failure. By the 1970’s, DARPA cut its budget for artificial intelligence, holding out longer than other governmental programs like the National Research Council which completely ended its funding of AI in 1966.
Thus began a period known as the AI winter, where research and public interest in artificial intelligence was at a low point. The hype of artificial intelligence was followed by disastrous public demonstrations of simplistic systems that far undershot expectations. Perhaps this was just a symptom of the Cold War; the Department of Defense may have believed that the Soviet Union thinking we have super powerful artificial intelligence is more important than real, organic progress in the sector. Regardless, growth lay dormant for decades.
But some gritty scientists kept searching for answers to the drawbacks of the perceptron. One of those was John Hopfield, a professor at CalTech and Princeton University, who studied physics, molecular biology, and neuroscience. Inspired by studying the way magnetic spins of materials called “spin glass” can reach chaotic low-energy states, and how our brain, containing billions of simplistic individual neurons, can create our mind, he sought to expand the capabilities of the now-humbled perceptron.
His very first paper in the field of neuroscience debuted the structure of the Hopfield network. In this network and unlike in its predecessor, the feed-forward neural network, the individual perceptrons point back to each other. This creates recursion, allowing inputs to loop through the network multiple times. And these Hopfield networks are able to store information as a memory system, similar to how (we think!) our brain stores memories. Hopfield networks proved much more effective at pattern recognition than the perceptron networks before them.

{kind=link}
Simultaneously, Geoffrey Hinton was studying machine learning algorithms at Carnegie-Mellon University when he detailed a network representing a community of neurons that can quickly learn underlying data patterns. The proposed “Boltzmann machines” help neural networks get to their solution faster.
The Hopfield network and the Boltzmann machine laid the groundwork for modern artificial intelligence systems by enabling neural networks to more quickly find complex patterns in their underlying data. These tools enabled researchers to design tailor-made neural networks to solve their problem of interest, like convolutional neural networks to understand images, or transformer neural networks to read lines of text. Without them, 1980’s computers would not have been able to solve these complex problems. Now, massive data centers can take advantage of increasing computing power and training data to teach these networks how to recognize patterns without relying on Hopfield networks or Boltzmann machines. But undoubtedly these discoveries enabled artificial intelligence research to get to this point at all.
Following the papers that penned the Hopfield network and Boltzmann machine, interest in artificial intelligence grew and the sector awakened from its AI winter. Hinton himself is a prolific author of papers in the AI field, contributing to numerous findings since that have earned him the title the “Godfather of AI”.
And today, the boom of artificial intelligence is as profound as ever. Hence the awarding of John Hopfield and Geoffrey Hinton with their Nobel Prize in physics.
Yet despite the clear impact of Hopfield and Hinton’s work, many people in physics communities have spoken against their Nobel Prize designation. I have seen numerous people claim their work was not physics-related, and while noteworthy in the fields of math and computer science, not applicable to the Nobel Prizez in physics. Some say that the Nobel Foundation is simply succumbing to the hype around AI.
I can certainly understand the disapointment of the Nobel Prize not going to a particular person or field that you may have a vested interest in. I am biased; I study bioengineering and computer science, so these awards are right up my alley!
But the Nobel Foundation was established in 1900 to give prizes in the fields of Physics, Chemistry, Physiology or Medicine, Literature, and Peace. At the time there was no inclusion of computer science or artificial intelligence because it didn’t exist!
From Hinton, in The New York Times:
If there was a Nobel Prize for computer science, our work would clearly be more appropriate for that. But there isn’t one.
The Nobel Foundation, established by Alfred Nobel in his last will and testament, was dedicated to acknowledging the discovery or invention of the previous year with the “greatest benefit on mankind”. Undoubtedly the creation of increasingly advanced artificial intelligence is an enormous benefit to humanity, with applications ranging from more accurate prediction of weather events to rapid medical diagnostic tests.
Physics is the bedrock of the other hard sciences. Physicists seek to understand how the world works, whether on an atomic scale or a planetary one. Both Hopfield and Hinton applied their previous work studying condensed matter physics and neuroscience to try and understand how our brains process information, and apply that to a general principle that governs memory. They are doing as physicists do best; identify and solve the root cause of a natural phenomena in as general terms as possible. And so the Nobel Prize of physics was the most natural award for John Hopfield and Geoffrey Hinton to win.
What is physics? To me—growing up with a father and mother who were both physicists—physics was not subject matter. The atom, the troposphere, the nucleus, a piece of glass, the washing machine, my bicycle, the phonograph, a magnet—these were all incidentally the subject matter. The central idea was that the world is understandable, that you should be able to take anything apart, understand the relationships between its constituents, do experiments, and on that basis be able to develop a quantitative understanding of its behavior. Physics was a point of view that the world around us is, with effort, ingenuity, and adequate resources, understandable in a predictive and reasonably quantitative fashion. Being a physicist is a dedication to the quest for this kind of understanding.
[...] I am gratified that many—perhaps most—physicists now view the physics of complex systems in general, and biological physics in particular, as members of the family. Physics is a point of view about the world.
Congratulations to all the Nobel Prize winners. And thank you for reading.